



胶质瘤是常见的脑瘤,按恶性程度分低级别胶质瘤和高级别胶质瘤。脊索样胶质瘤是一种良性、少见、生长缓慢的原发性中枢神经系统神经上皮肿瘤。国际卫生组织将它归类为WHO-2级肿瘤。脊索样胶质瘤的命名,在2000年就已经完成。在2021年的五版分类中,脊索样胶质瘤被归属于局限性星形胶质细胞瘤。脊索样胶质瘤,常常位于三脑室前部。那脊索样胶质瘤有哪些症状?发病率高吗?应该如何治疗?
上期精彩:【INC脑博士少见神外疾病必知系列八】下丘脑错构瘤
什么是脊索样胶质瘤?
脊索样胶质瘤少见,是一种良性且生长幻一的中枢神经系统上皮肿瘤,起源于胶质细胞,肿瘤边界清楚。常发生于三脑室前部或脊髓,组织学特征为表达GFAP的上皮样细胞的簇状和条索状排列,并且显示出PRKCA基因上p.D463H错义突变的重复出现(CNS WHO 2级)。ICD-O编码:9444/1脊索样胶质瘤。
脊索样胶质瘤有哪些常见症状?
因其位于三脑室,患者常出现脑积水、内分泌异常和/或视野缺损的症状,其他常见也包括疼痛、肢体无力、感觉异常、肌肉萎缩等。
脊索样胶质瘤组织病理学有何特点?
脊索样胶质瘤是一种实体肿瘤,常由不同黏液性间质内的上皮样细胞簇和条索组成。已报道三种不太常见的组织学模式:具有成片多边形上皮样细胞且无明显黏液基质的实性模式、松散胶原中具有成群梭形细胞的梭形模式和具有丰富胶原化的纤维化模式。纤维化模式在老年患者中更常见。单个肿瘤细胞具有丰富的嗜酸性细胞质。细胞核中等大小,卵圆形,相对均匀。核分裂像通常少见或不存在。间质淋巴浆细胞浸润,常含有大量Russell小体,是一种常见发现。与影像学表现一致,几乎没有脑浸润倾向。在邻近的非肿瘤组织中可见到反应性星形胶质细胞、Rosenthal纤维和慢性炎症细胞。
脊索样胶质瘤免疫表型有哪些特点?
研究显示脊索样胶质瘤显示GFAP的强的弥漫性表达,并持续表达转录因子TTF1(NKX2-1)。核TTF1染色的百分比和强度取决于使用的抗体克隆,少数病例显示表达少至不表达。波形蛋白和CD34的表达很强。S100和EMA的表达是可变的。神经元和神经内分泌标志物(突触素、神经丝蛋白、嗜铬粒蛋白A)始终为阴性。Ki-67增殖指数通常<2%。
脊索样胶质瘤诊断分子病理学有哪些研究进展?
目前研究显示,PRKCA基因的p.D463H错义突变几乎普遍存在于脊索样胶质瘤中,迄今为止已在研究的29个肿瘤中的28个中发现。尽管PRKCA基因在乳头状胶质神经元瘤中参与了基因融合,但尚未在任何其他人类肿瘤中发现该突变。因此,PRKCA p.D463H突变是诊断标志。脊索样神经胶质瘤缺乏其他脑肿瘤实体特征性基因的伴随致病性改变(例如,IDH1、IDH2、H3-3A、H3C2[_HIST1H3B]、_FGFR1、BRAF、NF1、CDKN2A、TP53)。还发现了脊索样胶质瘤独特的表观遗传特征,DNA甲基化分析代表了诊断确认的辅助方法。
脊索样胶质瘤的发病机制是什么?
目前脊索样胶质瘤的发病机制尚不清楚,可能与遗传、环境等因素有关。两项独自研究确定了影响PRKCA基因密码子463的新错义突变为分子标志改变。PRKCA编码PKC的催化α亚基,在多个跨膜受体下游的细胞内信号传导中起作用。尽管PRKCA在其他癌症中偶尔发生突变,但迄今为止尚未在其他人类瘤种中报告这种特异性p.D463H突变。该突变导致激酶结构域活性部位内密码子463处的组氨酸被天冬氨酸取代,其中天冬氨酸的侧链在ATP水解反应期间通常作为质子受体发挥作用。该突变的精确致癌机制仍有待阐明,但该突变可能改变底物特异性或催化活性{29476136;29915258}。已发现高水平的磷酸化ERK,表明PRKCA p.D463H突变可能至少部分通过激活MAPK信号通路发挥作用。
细胞起源:根据解剖位置、甲状腺转录因子1(TTF1)的一致免疫反应性和室管膜瘤样超微结构特征,脊索样胶质瘤被假设起源于终板血管器官的特化伸长细胞型室管膜细胞。
脊索样胶质瘤的发病率有多高?
脊索样胶质瘤的发病率较低,约占全部脊髓肿瘤的5-10%。该肿瘤多见于成人,平均年龄46岁,男女比例为1:2。
脊索样胶质瘤和其他类型的胶质瘤有什么区别?
脊索样胶质瘤主要发生在三脑室、脊髓,与其他胶质瘤的病理特点、临床表现和预后等方面有所不同。
脊索样胶质瘤的影像有哪些特点?
诊断方法主要包括磁共振成像(MRI)和活检术是确诊的主要依据,MR显示通常位于前三脑室内边界清楚的卵圆形或分叶状的肿块。T1与脑实质等强度,增强后明显均匀强化。受压相邻中枢神经结构(包括视神经束、基底节和内囊)可出现血管源性水肿,增强后明显强化,瘤周“八字水肿征”。
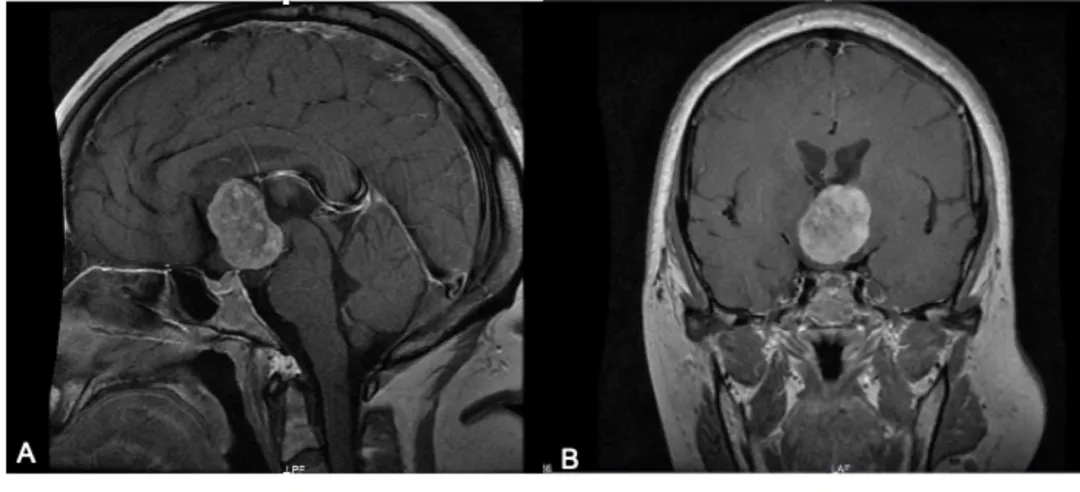
头颅MRI增强扫描示鞍上占位,边界较清。T1WI呈等低信号,T2WI呈不均匀等高信号,增强后病灶呈不均匀强化,邻近侧脑室、三脑室、视神经受压。A示增强矢状位,B示增强冠状位。
脊索样胶质瘤的分期是如何进行的?
目前尚无统一的脊索样胶质瘤分期方法,通常根据肿瘤的大小、侵犯范围和病理类型进行判断。
脊索样胶质瘤病理学上有何特别?
在病理学检查上,存在神经胶质抗原染色的索状和簇状上皮样细胞。大多数病例存在PRKCA基因的错义突变。不存在高级别肿瘤特征。
该疾病和哪些疾病需相鉴别?
和生殖细胞瘤鉴别要点:(1)鞍区生殖细胞瘤多见于儿童,女性多见,90%患者出现尿症;(2)T1WI/T2WI上多为等信号,边界相对比较清楚;可见于三脑室、鞍上、及鞍内,位于三脑室者多体大,出现坏死、囊变,呈明显不均匀强化。
和视神经胶质瘤鉴别要点:(1)常发生于10岁以内儿童;(2)视神经呈梭形、管状或球状增粗,边缘清楚;(3)增强后增粗的视神经呈轻度至明显强化。
和脑膜瘤鉴别要点:(1)肿瘤以鞍结节、前床突、后床突为中心向周围匍行生长,形成特征性的“蕈伞样”外观;(2)平扫等T1等T2信号;(3)增强后明显均匀强化,可有脑膜尾征。
和实性颅咽管瘤鉴别要点:(1)肿瘤呈不均匀等T1长T2,形状不规则;(2)增强后明显不均匀强化,内见微囊状不强化区。
和鞍区毛细胞型星形细胞瘤:鉴别要点:(1)多见于青少年;不规则分叶状肿块,边界清楚,体大;(2)增强后明显均匀强化。
脊索样胶质瘤的治疗方案有哪些?
治疗方案包括手术切除、放疗和化疗。
手术治疗脊索样胶质瘤的效果如何?
手术切除是脊索样胶质瘤的优选治疗方法,尽量保留患者的神经功能。手术效果因个体差异而异。肉眼下全切可治愈该病。
放疗和化疗在脊索样胶质瘤治疗中的应用和效果如何?
放疗和化疗常作为辅助治疗方法,用于手术后或无法手术的新兴治疗方法(如免疫治疗、靶向治疗等),放疗或立体定向放射外科对于不完全切除的病灶可能具有作用。肿瘤及其治疗的并发症可能包括尿崩症和记忆障碍。
在脊索样胶质瘤治疗中的应用前景如何?
新兴治疗方法尚处于研究阶段,尚无明确的临床数据支持其在脊索样胶质瘤治疗中的应用。但随着科学研究的深入,未来可能为脊索样胶质瘤治疗提供新的选择。
脊索样胶质瘤的预后因素有哪些?
预后因素包括肿瘤分期、病理类型、手术切除情况、放化疗效果等。
脊索样胶质瘤患者的生活质量如何?
生活质量与肿瘤侵犯范围、治疗效果等因素有关。治疗和康复训练可提高患者的生活质量。
脊索样胶质瘤患者应该注意哪些生活习惯的改变?
患者应保持良好的作息、饮食习惯,避免劳累、情绪波动,定期进行复查和康复训练。
脊索样胶质瘤患者在康复期需要注意什么?
康复期患者应按医嘱进行物理治疗、康复训练,定期复查,及时发现并处理并发症。
脊索样胶质瘤复发的原因有哪些?
复发的原因可能与肿瘤分期、病理类型、手术切除情况等因素有关。
如何预防脊索样胶质瘤的复发?
尽量争取完全的根治性治疗是预防复发的关键独自性的因素,经治疗后预防复发的方法包括定期复查、严格遵循医嘱进行康复训练和治疗。
小结
综上所述,脊索样胶质瘤虽然是WHO-2级局限性的胶质瘤。但是因为生长的位置位于大脑深处的下丘脑处,手术难度很大,全切除后能长久不复发。脊索样胶质瘤的治疗是基于大水平的肿瘤切除,同时避免并发症,如尿崩症和其他内分泌功能障碍。
