



神经鞘肿瘤主要由神经鞘瘤和神经纤维瘤组成,是成人二常见的原发性脊髓肿瘤。在美国,这些肿瘤的发病率估计为每10万人中有0.26例,男性的发病率略高(优势比,1.11)。这些肿瘤中超过大概率是良性的,但恶性神经鞘肿瘤虽然少见,但却是一个独特的、众所周知的难以治疗的子集,具有很高的发病率和死亡率。
遗传学
虽然这些肿瘤大多偶尔发生,但已知神经纤维瘤和神经鞘瘤与1型神经纤维瘤病(NF1)和神经系统功能障碍有关NF1肿瘤控制基因,以及与NF2和损失NF2基因功能,进而产生支架蛋白灰背隼。脊柱肿瘤在NF2很常见,据估计超过67%的患者的脊柱肿瘤可以通过核磁共振成像检测出来。神经鞘瘤可以在NF1患者中发现,同样神经纤维瘤也可以在NF2环境中见到。此外,肿瘤存在混合或混合病理,难以在病理学上明确分类为神经鞘瘤或神经纤维瘤。虽然不常见,但当遇到这些肿瘤时,很可能是潜在的遗传疾病。
三种主要的神经纤维瘤病综合征,神经鞘瘤病,最近被发现。虽然这种综合征的确切定义和诊断标准仍在发展中,但对这种综合征的认识承认了这样的观察,即许多患者表现为多发性神经鞘瘤或不符合NF2诊断标准的家族性神经鞘瘤。这SMARCB1在22号染色体上发现的肿瘤控制基因与染色质重塑有关,它与许多这类病例有关,高达45%的家族性神经鞘瘤病例被发现具有SMARCB1突变。
脊髓神经鞘瘤治疗
外科手术
与神经纤维瘤病无关的神经鞘肿瘤的治疗传统上包括较大限度的顺利手术切除,尽管可以观察到某些具有无症状病变的患者的生长或症状/体征发展(图1)。如果肿瘤的近端和远端被识别,并且周围的神经根从肿瘤壁上脱离,则完全切除通常是可行的。牺牲1-2个被肿瘤包裹的神经根是可以忍受的,并且神经功能缺损较小。对于神经鞘瘤来说,完全切除是典型的,但是神经纤维瘤更难切除,因为涉及到神经根和纤维。
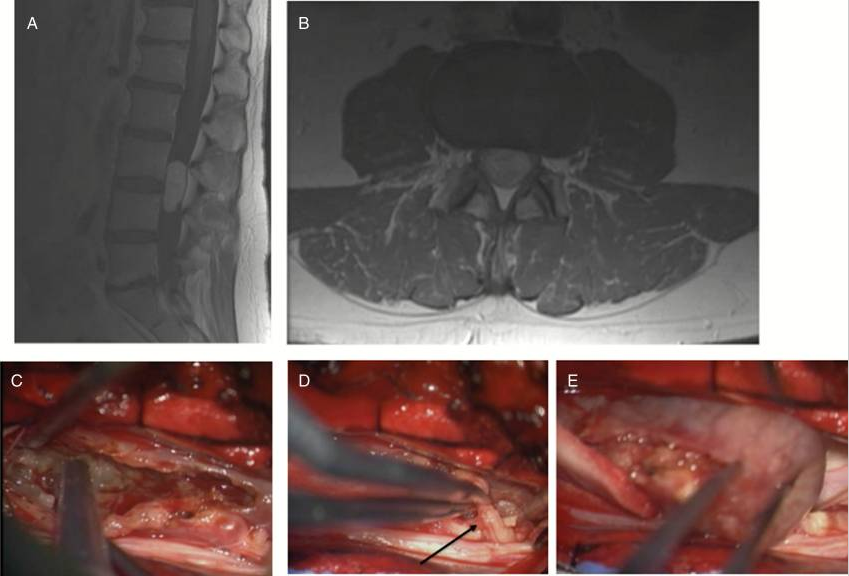
图:神经鞘瘤。一名44岁男性,右下肢疼痛和麻木数月。(A,B)MRI显示大的硬膜内肿瘤。(C) 用超声吸引器清除肿瘤。(D) 发现一个神经根与肿瘤有关,在刺激下,没有引起运动反应,可以牺牲神经根(箭头)。(E) 肿瘤可以完全切除。
放射治疗
最近无框架立体定位精度的提高导致人们对放射外科和大分割放射在一般脊柱肿瘤和特别是神经鞘肿瘤治疗中的应用越来越感兴趣。申等人研究了立体定向放射外科在66例脊神经鞘肿瘤患者中的应用。平均随访44个月,他们报告局部控制率为95.4%,视觉模拟疼痛评分有好转,没有报告并发症或辐射诱导的神经毒性。其他几个病例系列也报告了类似的结果,全部病例都具有很高的局部控制率,很少或没有报告神经毒性。然而,应该注意的是,这些研究者取得的有希望的结果仍然有局限性,较突出的是在随访期间。由于神经鞘肿瘤通常生长缓慢,较短的随访时间可能会错过许多治疗失败或延迟恶性转化的病例。
系统疗法
作为生长缓慢的肿瘤,脊神经鞘肿瘤通常被认为对标准化疗方案没有反应。然而,研究人员继续探索神经纤维瘤病患者的可能药物治疗,特别是预防丛状神经纤维瘤和不可切除肿瘤的进展。TKI伊马替尼已经在二阶段临床试验中进行了测试,并显示出一些益处。26%接受研究药物的患者出现丛状神经纤维瘤体积减少≥20%。mTOR控制剂西罗莫司(雷帕霉素)在对46名患者的一项二期临床试验中,也显示了一些适度的益处。服用西罗莫司的患者神经纤维瘤的进展时间较慢,平均为4个月。最近,丝裂原活化蛋白激酶的控制剂selumetinib也显示出前景。24名儿童最近接受了一期试验治疗,71%获得部分缓解(肿瘤体积减少3% -20%),0%报告疾病进展,大多数儿童能够耐受延长的治疗方案。这可能是其中一些治疗将合适对抗椎管内神经纤维瘤。
其他化疗方案和分子靶点也已经过测试,但显示的益处有限。Tipifarnib是一种法呢基转移酶控制剂,已被用于控制NF1患者丛状神经纤维瘤中出现的失调的Ras信号通路。尽管最初的一期试验数据表明该药物在儿科人群中耐受性良好,但随后的二期试验未能证明丛状神经纤维瘤的进展时间有所好转。
很少有试验研究NF2相关神经鞘瘤的药物治疗,但迄今为止,在评估靶向表皮生长因子受体(EGFR)活性的药物时,成功有限。拉帕替尼是一种选择性EGFR控制剂,已被证明对室管膜瘤有活性,但后续结果喜忧参半。
脊髓神经鞘瘤预后如何?
如果肿瘤可以在不损伤潜在神经的情况下切除,脊髓神经鞘瘤(良性肿瘤)的预后是较好的。然而,这也取决于脊髓上的肿瘤部位以及相关体征和症状的严重程度。无症状的肿瘤可能不需要治疗;除非它们引起不适、影响生活质量或影响潜在神经的功能。在这种情况下,密切监测神经鞘瘤是一种治疗选择。如果良性神经鞘瘤转化为恶性肿瘤(在少见的情况下),那么预后可能取决于一系列因素,包括肿瘤的阶段、在脊柱上的位置、个体的整体健康状况以及对治疗的反应
