髓母细胞瘤约占儿童中枢神经系统肿瘤的20%,成人仅仅占了1%。在70–80%的病例中,髓母细胞瘤影响16岁和16岁以下的儿童。平均发病率在每100,000人0.20至0.58例之间。
目前,5年总生存率(OS)达到70%,但患者的预后差异较大。根据年龄、转移情况、术后残留的程度以及肿瘤的组织学特征,将髓母细胞瘤分为标危和高危。标危和高危髓母细胞瘤的5年生存率分别超过80%和60%。
对于标危患者和高危患者,常规治疗包括较大切除手术,颅脑脊髓照射(CSI)和化学治疗(CT)的结合。这种组合可在60-80%的患者中实现长期OS,但往往以长期毒性为代价。因此,对疾病的更好的了解似乎是一个难题,目的是提供更准确的风险适应性治疗并开发针对性的疗法,以减少低危患者的副作用并提高高危患者的生存率。在过去的二十年中,在了解髓母细胞瘤的分子机制方面已经取得了进展。这些已经达成国际共识,以定义髓母细胞瘤的预后分子亚组,其已被包括在最近出版的经修订的WHO《中枢神经系统肿瘤分类》四版中。由于针对亚组的临床试验方法的发展,对髓母细胞瘤的分子理解已开始转化为临床应用,从而可以更准确地分配放射剂量或化疗方案,并评估新兴靶向治疗的效率。
分类
目前的预后分类将髓母细胞瘤分为“标准风险”和“”,其依据是年龄、转移情况、术后残留的程度和组织学。5种组织学亚型分别是:经典型、促纤维增生性/结节型、广泛结节型、间变型和大细胞型。大细胞和间变型髓母细胞瘤有相似的细胞学特征,因此,这些类型常常被认为是“大细胞/间变型组织学”,预后不良。大细胞髓母细胞瘤是由具有单个核仁的均匀的大圆细胞组成,其有丝分裂和凋亡指数也高于其他组织。广泛结节型和促纤维增生型预后较好。术后残留病灶少于1.5 cm 2的患者预后较好。转移的判定采用Chang标准,区分4种转移水平:脑脊液(CSF)中有肿瘤细胞(M1);颅内结节状种植(M2);脊髓结节状种植(M3);颅外转移(M4)。婴儿(3岁以下)的存活率低于年龄较大的儿童,有可能是治疗策略的问题,不能放疗造成的结果。
因此,根据该临床和组织学分类,标危患者大约占髓母细胞瘤的70%大部分是年龄>3岁的儿童,在颅脊髓磁共振成像或CSF细胞学检查中没有发现肿瘤细胞,术后残留小于1.5 cm 2和非大细胞/间变型组织学。五年控制率达到85%。没有以上标准的则归为高危患者,五年总体生存率下降到约60%。
分子分型
2002年,Pomeroy等人提出了髓母细胞瘤不同分子亚群。他们通过研究DNA微阵列基因表达,数据显示,PTCH,GLI和MYCN(SHH的全部三个转录靶标)与增生性/结节性髓母细胞瘤高度相关。有几项研究开始根据转录组的差异对髓母细胞瘤进行分类,并得出了大致一致的结论。Thompson等人2006年,以及Kool等人2008年,总结了A、B、C、D和E.Cho等五种不同的分子亚型的存在。2011年得出结论,存在六种不同的分子亚型,分别命名为C1到C6和Northcott等。2011年得出结论,存在四种不同的分子亚型,即SHH、WNT、C组和D组。2010年秋季在波士顿举行了一次会议,会上一致认为髓母细胞瘤有四个主要的转录亚组,分别命名为WNT、SHH、Group 3和Group 4,在人口学、组织学、DNA拷贝数变异和临床结果方面明显不同。
WNT亚型是较少见的(占全部髓母细胞瘤的10%),但有较好的临床预后,5年OS>95%。这种分子亚群主要与经典组织学有关。WNT髓母细胞瘤很少有大细胞/间变性组织学,但即使是这种组织学,它们也有很好的预后。WNT髓母细胞瘤发生于3岁以上的儿童或青少年。分子机制由WNT信号通路的激活来定义,通过β-catenin的表达作为一种信号转导来实现的。在85-90%的病例中,wnt信号通路的激活是由于激活了wnt3外显子的体细胞突变。CTNNB 1导致β-catenin过度表达。6号染色体缺失在WNT肿瘤中也有较高的复发率,占70%-80%.较少发生TP 53突变,SMARCA 4,和DDX3X突变。Wnt髓母细胞瘤很少携带APC与Turcot综合征。在WNT髓母细胞瘤的少见病例中也发现了ALK的突变,其生理病理学尚不清楚。
SHH亚群占全部髓母细胞的30%,预后中等,5年OS为70%.促纤维增生性/结节性组织学与SHH肿瘤有很强的相关性,因为绝大部分促纤维增生性/结节型属于SHH亚型,但是大约有50%的SHH亚型髓母细胞瘤不是促纤维增生性/结节性。Shh髓母细胞瘤较常见于婴儿和成人,而在4-15岁的患者中很少见。这些肿瘤来源于外颗粒层的小脑颗粒前体细胞。其分子机制涉及SHH信号通路的过度表达。
通过PTCH1,SMO,GLI和SUFU作为一种转导增强剂。SHH通路的遗传事件与年龄有关:在婴儿中,PTCH1(格林综合症)或SUFU的种系突变很常见。所以,在Gorlin综合征患者中,应避免放疗,因为放疗会引起继发性癌症(主要是
脑膜瘤和基底细胞癌)的风险。此外,即使仅采用化疗方案,患有SHH髓母细胞瘤的婴儿也有很好的预后.3至16岁的儿童大多表现出PTCH1的突变或TP53的突变(李·弗劳梅尼综合症)因此,全部的小儿SHH肿瘤都应做遗传易感基因筛查来排除Gorlin综合征,Li Fraumeni综合征,或SUFU突变。
TP 53突变常与GLI 2和MYCN扩增同时存在,诱导SHH通路的激活。TP 53突变存在于~30%的SHH髓母细胞瘤中,预后较差,5年OS为40%。因此,国际卫生组织《中枢神经系统肿瘤分类》四版将具有或不具有TP53突变的SHH髓母细胞瘤分开。众所周知,表达TP 53突变的放射敏感度较低,Tchelebi等人提示放疗甚至可以促进髓母细胞瘤的生长。在成年人中,较常见的突变是PTCH1,SMO,以及TERT启动子突变,或偶尔在在IDH 1突变。
3组髓母细胞瘤占全部髓母细胞瘤的25%,预后较差,儿童5年生存率为58%。而未放疗婴儿的OS更差(5年OS为45%)。肿瘤以经典组织学为主,但这一组的大细胞/间变性组织学比率也很高(40%),是在婴儿中。3组髓母细胞瘤多发生于男性(2:1)和16岁以下者,来源于小脑干细胞。与WNT和SHH亚组不同,在WNT和SHH亚组中,一个功能失调的分子通路已经被明确地识别出来,但在3组髓母细胞瘤中没有明确的定义。MYC扩增(3组10%至20%),17q以及MYC扩增的预后特别差,五年OS为20%。3组髓母细胞瘤具有很高的转移扩散力,因为40%–45%的患者在诊断时具有软脑膜扩散,并且复发后大多带有转移性。
4组髓母细胞瘤,虽然是较常见的(35%的全部髓母细胞瘤),是较不了解的分型。该亚组主要表现为经典的组织学,发生在全部年龄,主要以男性为主(3:1)。4组髓母细胞瘤临床疗效中等,5年生存率为75~90%,但是对于不能从放疗中受益的婴儿来说,这是很差的。总体上,30%-40%的4组患者诊断为转移。对于4组,没有明确的原因。等染色体17q在4组肿瘤中多见(丢失17 p63%,增益17q73%)。虽然,与3组肿瘤不同,这种异常并不会给这个亚组带来不良的预后。MYCN或CDK 6扩增,SNCAIP复制,X染色体缺失,和失活的KDM6A(占4组的10%),7号染色体扩增(47%),8号染色体缺失(41%),10Q缺失(15%),12Q扩增(20%),18号染色体增益(16%),并11号染色体缺失,是一个较好的预后指标。
因此,尽管分子亚组已经完全改变了髓母细胞瘤的分类,但是亚组内存在很大的异质性,还需要进行大量研究来提高对髓母细胞瘤的分子理解。
最近的研究建议更准确的分子筛选。髓母细胞瘤亚组的更好的特征对3组和4组尤为重要,因为如上所述,3组和4组髓母细胞瘤的转录序列是相似的,两组中都有一些细胞遗传学特征,如同染色体17q(I17q)。然而,3组和4组的结果有所不同,是转移扩散的趋势。2017年,通过使用相似网络融合方法分析基因组范围的dna甲基化和基因表达数据,Cavalli等人。提示每个亚组均存在与临床和生物学相关的亚型。
他们的结论是,这四个分子亚组可进一步分为12个不同的亚型,在分子、临床和预后方面存在差异(WNT:2,SHH:4,3:3,4:3)。同样,2017年,Northcott等人分析了491个测序的髓母细胞瘤样品的体细胞结构以及1,256个遗传学分析病例中的分子异质性,发现了新的肿瘤亚型,是3组和4组。2017,Schwalbe等对428例原发性髓母细胞瘤样品进行了分子谱分析,包括DNA甲基化微阵列分析,并鉴定了儿童髓母细胞瘤的七个分子亚组(WNT亚组保持不变,每个剩余的共有亚组被分为两部分)。这些数据有望好转疾病风险分层并根据其基因型治疗患者亚型。
1、手术治疗的目的在于尽可能地切除肿瘤,获得病理诊断,减轻颅内占位效应,降低颅高压,但是单纯手术不能根治髓母细胞瘤。
2.手术治疗+放疗是髓母细胞瘤治疗的标准模式,全脑全脊髓是放疗的靶区,尽量在术后30天内开始放疗。
3.化疗的目的是为了延长总生存期,提高患儿的生存质量和生活质量,但是仅靠单纯化疗,冶愈髓母细胞瘤的机会小,也就是说放疗才是髓母细胞瘤不可缺少的治疗手段。放疗联合化疗是可行的,但要优先保障放疗的完成,低剂量照射或放疗中断会影响总生存期。
INC国际Rukta教授的治疗案例
一名8岁男童因持续的头痛、呕吐就诊,起初家人认为只是疲劳和感冒造成的小问题,于是为其请假休养,但病情并未好转,甚至持续了三个月,患儿还出现了躯干性共济失调,走路不稳、举止异常笨拙,此时家人才发觉事态严重,就医后患儿被诊断为髓母细胞瘤,但由于病情凶险、位置深在,医院委婉建议转院、由更有经验的医生治疗。患儿家属查阅了很多论文和期刊后,决心前往INC国际专家、国际神经外科杂志《Journal of Neurosurgery》主编James T. Rutka教授所在的SickKids医院,并希望由Rutka教授为其制定综合治疗方案以延长患儿的生存期。
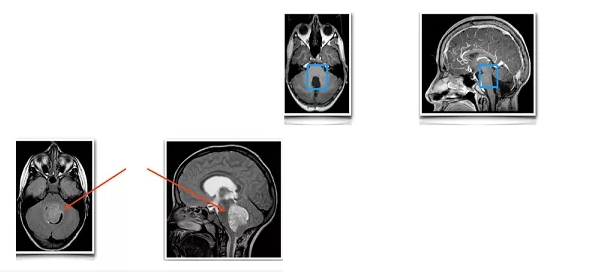
得了髓母细胞瘤能活多久?
Rutka教授仔细研究了患儿的病历资料后决定先行手术近全切肿瘤,随后辅以颅脊髓照射和周期性化疗,患儿对此耐受良好,并且带瘤生存长达8年之久,随访显示其顺利入学,虽然仍在接受后续治疗,但患儿一家认为“他已经足够幸运,感谢Rutka教授挽救了孩子的生命”。此外,Rutka教授还曾在TED演讲中交流过他的“奇迹”患者、“抗癌大使”朱利安的故事,他的治疗案例也鼓舞了其他脑瘤患儿及其家属,重拾治疗信心。
后记
James T. Rutka教授曾在《The molecular genetics of medulloblastoma: an assessment of newtherapeutic targets》这份研究中描述了目前关于髓母细胞瘤基因状态的研究策略,提出今后治疗髓母细胞瘤的新希望和新方法,例如下代基因测序以及干细胞治疗等。同时,教授强调:医学研究者需要深入了解髓母细胞瘤的分子生物学,以开发针对这种肿瘤的新型治疗药物。值得一提的是,Rutka教授还较为擅长利用国际前沿技术激光间质热疗(LITT)治疗小儿脑瘤、癫痫等,既能使患儿免受开颅手术之苦,又能从根本上消融病变、提高生存质量,越来越多的发达国家患者也选择接受此技术治疗脑深部病变、复发性及恶性脑肿瘤、癫痫以及放射性坏死等。




