



胶质瘤分子病理诊断共识更新
脑胶质瘤确诊需要通过肿瘤切除手术或活检手术获取标本,进行组织病理和分子病理整合诊断,确定病理分级和分子亚型。分子标志物对脑胶质瘤的个体化诊疗及临床预后判断具有重要价值。脑胶质瘤治疗以手术切除为主,结合放疗、化疗等综合治疗方法。手术可以缓解临床症状,延长生存期,并获得足够肿瘤标本用以明确组织病理学和分子病理学诊断。随着病理学的发展和病理检测技术的进步,尤其是二代测序、DNA甲基化谱等组学技术的提高,胶质瘤的遗传背景和发生发展机制逐渐清晰。越来越多的分子标志物被证明在胶质瘤的分类、分型、分级、预后和治疗方面发挥着重要的作用,那目前诊断指南中,对于胶质瘤中具有诊断价值的基因和染色体改变呢?

2025年6月中华医学会病理学分会发表脑胶质瘤分子病理诊断专家共识,结合国内外研究进展及实践经验,就重要分子指标、诊断流程、技术优劣及检测路径进行阐述,提出了如下常见脑胶质瘤分子整合病理诊断流程,更加适合中国临床实践。
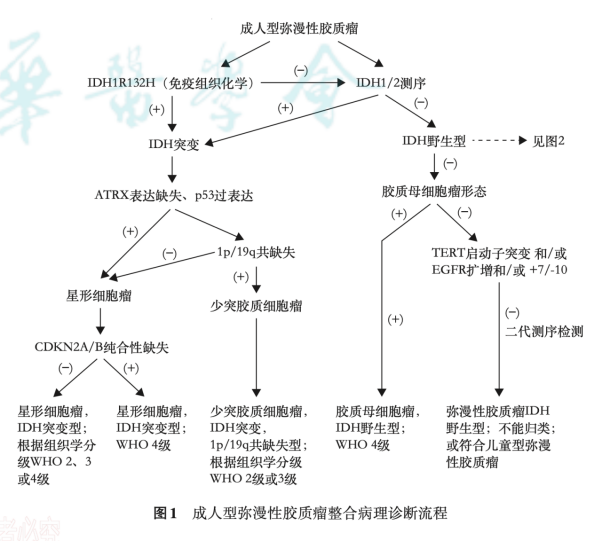
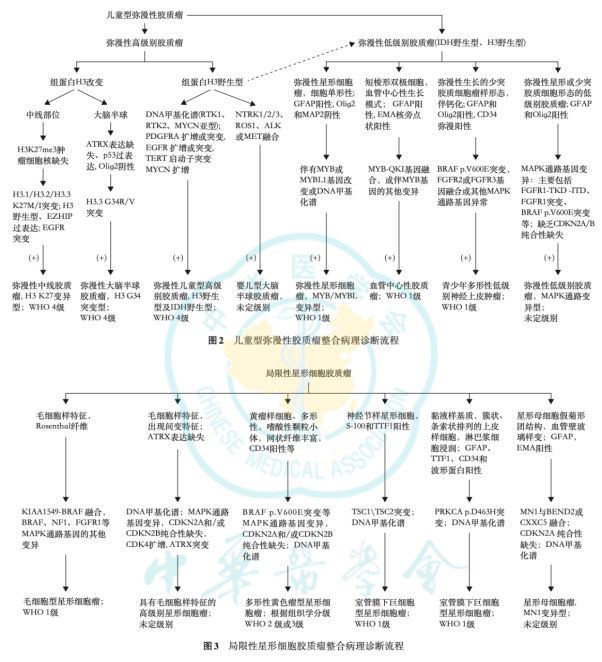
1. IDH1/2 基因突变
IDH (isocitratedehydrogenasegene)基因编码异柠檬酸脱氢酶,是三羧酸循环中起关键作用的酶家族之一。目前与胶质瘤相关的IDH基因有IDH1和IDH2两种亚型,主要见于IDH 突变型星形细胞瘤和IDH突变伴1p/19q共缺失型少突胶质细胞瘤中。IDH1和IDH2基因突变分别集中于编码IDH1蛋白第132位精氨酸残基(R132)的密码子(CGT)及编码IDH2蛋白第172位精氨酸残基(R172)的密码子(AGG)。
IDH1/2 基因突变在胶质瘤中的分布及临床意义

2. 染色体 1p/19q共缺失
1p/19q共缺失是第1号染色体长臂(1q)和第19号染色体短臂(19p)之间发生不平衡易位形成融合染色体1q/19p的结果,导致一条第1号染色体短臂(1p)和一条第19号染色体长臂(19q)同时发生完全性缺失,而另一条第1号染色体和另一条第19号染色体正常,故荧光原位杂交(FISH)仅能检测到单拷贝1p 和19q,所以1p/19q共缺失是杂合性缺失(LOH)。
1p/19q染色体臂的不完全或部分缺失不符合少突胶质细胞瘤的诊断标准,但可发生于IDH野生型胶质母细胞瘤病例。存在1p/19q共缺失和IDH突变的少突胶质细胞瘤生长速度较慢,并对丙卡巴肼+洛莫司汀+长春新碱(PCV)联合化疗和替莫唑胺化疗更加敏感,总生存期明显延长。
3.端粒酶逆转录酶(c)基因启动子突变
TERT可通过催化端粒复制维持其有效长度以促进细胞增殖。TERT基因启动子突变可持续激活TERT使肿瘤细胞获得无限增殖能力。胶质瘤中存在的TERT基因启动子突变,主要为C228T或C250T突变。
其中,IDH突变型胶质瘤,主要少突胶质细胞瘤,常伴有TERT基因启动子突变,可辅助其诊断;而成人型IDH野生型弥漫性胶质瘤存在TERT基因启动子突变应整合诊断IDH野生型胶质母细胞瘤(CNS WHO 4级),且预后较差。
4.CDKN2A/2B 基因纯合性缺失
CDKN2A和CDKN2B基因位于第9号染色体上,均为抑癌基因。CDKN2A基因编码p14M和p16^蛋白,与其相邻CDKN2B基因编码p15INK4b蛋白,三者均是重要的细胞周期素依赖激酶(CDK)抑制因子,它们均可通过抑制CDK活性阻止细胞周期G1的进程,从而抑制细胞增殖。CDKN2A/2B基因的纯合性缺失可导致细胞增殖失控乃至肿瘤发生。
在有IDH突变,且组织学表现符合2或3级星形细胞瘤者,如同时存在CDKN2A和/或CDKN2B 基因纯合性缺失,则要整合诊断为CNS WHO 4级的IDH突变型星形细胞瘤。此外,应注意CDKN2A未发生纯合性缺失是儿童型MAPK通路改变型弥漫性低级别胶质瘤的必要诊断标准之一。
5.表皮生长因子受体(EGFR)基因变异
EGFR基因定位于染色体7p12.编码一种跨膜酪氨酸激酶受体EGFR。在IDH野生型胶质母细胞瘤中约60%的肿瘤可见EGFR基因扩增、突变、重排或剪接改变,其中最常见的是扩增。在IDH野生的成人型弥漫性胶质瘤中,即使组织形态表现CNSWHO 2~3级,且无肾小球样血管增生和假栅栏样坏死,如出现EGFR基因扩增,则整合诊断IDH野生型胶质母细胞瘤(CNS WHO4级)。
20%~30%的胶质母细胞瘤可伴发EGFR基因截短突变(其外显子2~7框内缺失形成的EGFRvIII截短性重排),导致其编码缺少细胞外结构域的EGFRvIII截短蛋白,EGFRvIII胞内酪氨酸激酶结构域通过自发形成二聚体化和自磷酸化异常激活,持续活化下游信号转导,刺激肿瘤细胞增殖。此外,在弥漫性中线胶质瘤中可出现EGFR基因突变,多为编码细胞内酪氨酸激酶结构域的第20号外显子框内插入/复制,少数第7号外显子(编码细胞外结构域部分)的错义突变,多见于p.A289T = p.A289V.
6.BRAF基因变异
BRAF基因编码一种丝/苏氨酸特异性激酶,是RAS/RAF/MEK/ERK/MAPK通路重要的信号传递因子,介导细胞生长、增殖和分化。在胶质瘤中主要发生BRAF基因V600E错义突变和BRAF基因融合。BRAFp.V600E突变是指BRAF蛋白第600位缬氨酸残基(V)被谷氨酸残基替换(E),可发生在儿童型低级别弥漫性胶质瘤、节细胞胶质瘤、青少年多形性低级别神经上皮肿瘤、毛细胞型星形细胞瘤、多形性黄色瘤型星形细胞瘤及上皮样型胶质母细胞瘤中。
BRAF基因融合染色体7q34位点的串联重复引起BRAF-KIAA1549融合,该融合主要发生在毛细胞型星形细胞瘤中,也可见于弥漫性软脑膜胶质神经元肿瘤及少量儿童型弥漫性低级别胶质瘤和局限性星形细胞胶质瘤中。毛细胞型星形细胞瘤中另有一些少见的融合如BRAF-RNF130、BRAF-CLCN6等。
7.ATRX基因变异
ATRX基因X染色体连锁的a地中海贫血智力发育迟滞综合征(a-thalassemia/mental retardation syndrome X-linked)的致病基因。ATRX蛋白是一种ATP依赖的解旋酶,具有参与DNA修复、转录调节、维持端粒稳定和染色质重塑等广泛的生物学功能。在第5版WHO CNS肿瘤分类中,ATRX基因的状态亦是定义胶质瘤分子分型的关键标志之一。
ATRX基因的变异形式主要为失活变异,可能由ATRX基因的突变、缺失或融合等原因所引起,免疫组织化学染色显示肿瘤细胞不表达该核蛋白(内皮细胞核阳性可作内对照)。IDH突变的成人型弥漫性星形细胞瘤中,IDH1/2基因突变与ATRX基因失活突变和 TP53基因突变存在明显的相关性,而在少突胶质细胞瘤中,ATRX基因野生型。此外,ATRX 基因突变或蛋白表达缺失还常见于H3K27变异型弥漫性中线胶质瘤、H3G34突变型弥漫性大脑半球胶质瘤及具有毛细胞样特征的高级别星形细胞瘤中。
8.H3 K27突变
H3K27变异型弥漫性中线胶质瘤(diffuse midline gioma, DMG)最常见的分子改变是H3 p.K28M/I(K27M/I)突变,诊断弥漫中线胶质瘤的主要特征。H3 K27变异型DMG均出现组蛋白3(H3)中第28位赖氨酸残基(K28)三甲基化(me3)障碍,表现为细胞核广泛缺失K28三甲基化的H3(H3K28me3)。其中H3.3、H3.2或H3.1 p.K28M(K27M)突变使其作为 me3位点的K28被更换非me3位点的甲硫氨酸残基(M),直接导致H3K28me3 (H3K27me3)缺失。
H3 p.K28M(K27M)突变蛋白还可与甲基转移酶EZH2相互作用,进而抑制PRC2活性,也可导致H3K28me3 (H3K27me3)缺失。H3K28me3(H3K27me3)缺失通过影响所在部位染色质核小体DNA空间构象,促进癌基因转录和/或阻止抑癌基因转录,从而导致DMG的发生、发展。个别DMG病例为H3p.K28I(K27I)突变,H3p.K28I(K27I)突变是K28被异亮氨酸残基(I)取代,发挥与 K28M(K27M)类似的机制并引起 H3K28me3(H3K27me3)缺失导致DMG的发生、发展。
9.H3 G34突变
H3G34突变型弥漫性大脑半球胶质瘤 (diffuse hemispheric glioma,H3 G34-mutant, H3G34 DHG)是一组发生在大脑半球的弥漫浸润性胶质瘤,伴有H3.3(H3-3A,也称为H3F3A4: 1) p. G35 (G34) R/V FE. H3.3 p.G35(G34)R/V突变即其第35位甘氨酸残基(G)被精氨酸残基(R)或缬氨酸残基(V)替代,由此引起H3.3与SETD2 和KDM2A结合障碍,直接影响组蛋白3(H3)第37位赖氨酸残基[H3p.K37(K36)]的甲基化状态,并可阻止 H3p.K37(K36)与人错配识别蛋白MutSa 的相互作用,共同抑制H3中的K37 三甲基化(me3),而H3 p. K37(K36)me3 减少可通过上调原癌基因 MYCN 表达诱发 H3 G34 DHG。
参考资料:
1.胶质瘤分子病理诊断中国专家共识2025版.
2.WHO中枢神经系统肿瘤诊断分级.2021
