



神经节细胞胶质瘤是一种少见的、生长缓慢的原发性中枢神经系统(CNS)肿瘤,较常见于颞叶,通常会导致癫痫发作。脊髓神经节细胞胶质瘤较其少见,仅报道了大约70例脊髓神经节细胞胶质瘤,大多数发生在儿童或年轻人中。治疗的金标准是全切除。
相关阅读—INC德国巴特朗菲教授:关于小儿颅内原发性间变性神经节胶质瘤(AGG)
案例报告
一名14个月大的女婴最初被评估为颈部姿势异常,头部向右上方转动困难。病人被注意到执行补偿性动作,例如用她的手臂抬高自己环顾四周,而不是简单地抬起头。类似地,观察到患者向左转270°,而不是简单地向右转90°。体检发现头部向左偏,右侧胸锁乳突肌的体积和张力增加,右肩抬高。右侧胸椎侧凸也存在。试图移动脖子引起病人哭泣。就她的年龄而言,她有良好的细致运动协调能力,但还不能独自行走。没有出现额外的运动、感觉、步态或反射异常。脊柱侧凸的体检结果促使咨询到矫形外科,随后进行了核磁共振成像。
磁共振显示长节段髓内病变从T1延伸至T11水平,伴有相应的脊髓扩张,钆注射后出现不均匀强化【图1】、等强度T1信号和非均匀高强度T2信号【图2】。在增强肿瘤的头侧和尾侧有相关的脊髓空洞症。此外,在延髓和上颈椎以及脊髓尾部有T2高信号,无相关增强。
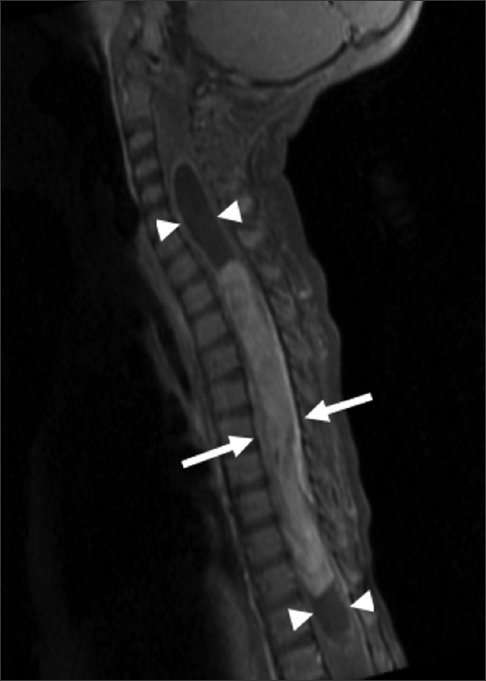
图1:14个月大的女童,患有脊髓神经节细胞胶质瘤。颈椎和胸椎矢状T1脂肪控制钆对比增强磁共振图像显示从T1延伸至T11的长节段增强髓内病变(箭头),增强肿瘤的头侧和尾部有脊髓扩张和脊髓空洞(箭头)。
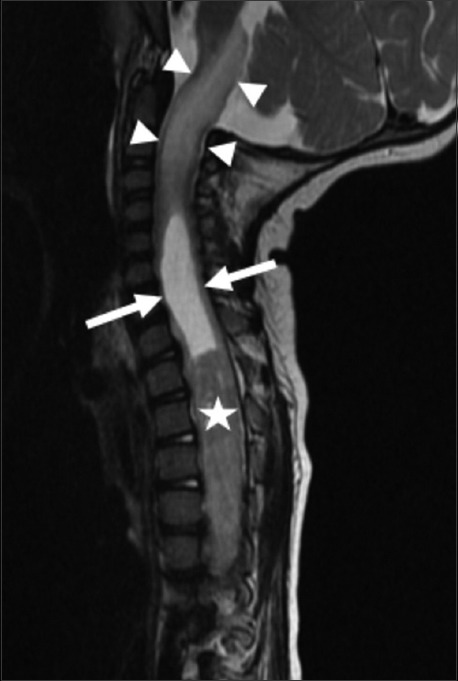
图2:14个月大的女童,患有脊髓神经节细胞胶质瘤。颈椎和胸椎的矢状T2加权磁共振图像显示脊髓空洞(箭头)位于髓内肿瘤(星形)的头侧。异常高强度T2信号和扩张见于上颈脊髓和脑干(箭头)。
患者随后接受了胸椎整块T1-T12椎板切除术,并使用肌电图、体感诱发电位、运动诱发电位和D波神经监测对胸髓内脊髓肿瘤进行了去瘤。使用后脊髓切开术和腔管超声外科吸引器(CUSA)的组合来去除肿瘤。不幸的是,在探查过程中,发现肿瘤与正常脊髓之间有不同程度的穿插。因此,只有次全切除才能顺利实现,术中估计约60-70%的肿瘤切除。
术后,患者保持了全部肢体的正常功能,磁共振显示增强肿瘤的大小减小,脊髓扩张减少,邻近脊髓的T2高信号增强。然而,由于较大的残留肿瘤负担,开始用卡铂和长春新碱进行化疗。不幸的是,3个月后随访成像显示髓内肿瘤增强,脊髓扩张恶化,T2高信号。化疗改为长春碱。尽管有疾病进展的影像学证据,患者没有表现出新的神经功能缺损,尽管脊柱侧凸和异常的颈部姿势仍然存在。在开始使用长春碱后6个月的额外随访磁共振显示脊髓水肿好转,肿瘤增强减弱。在撰写本文时,患者能够在无人帮助的情况下行走和跳跃,已经开始说话,并继续恢复。
手术切除的切片显示弥漫性胶质原纤维酸性蛋白(GFAP)免疫反应性神经胶质瘤,聚集了不典型的、可变的双核突触素免疫反应性细胞,没有神经或神经丝免疫反应性【图3–5】。有局灶性坏死和钙化,但无嗜酸性颗粒体。异柠檬酸脱氢酶基因1 (R132)或p53无免疫反应。分化簇34 (CD34)免疫染色仅限于血管。网状蛋白无明显的细胞周染色。有少见的有丝分裂,但Ki-67标记是可变的,局部为15%。聚合酶链反应显示没有BRAF v600E突变,NMYC或表皮生长因子受体基因扩增,和荧光就地杂交没有发现染色体10q丢失。对神经胶质细胞瘤的诊断符合国际卫生组织ⅰ级神经节细胞胶质瘤。这一诊断得到了来自不同学术机构的二种意见的证实。
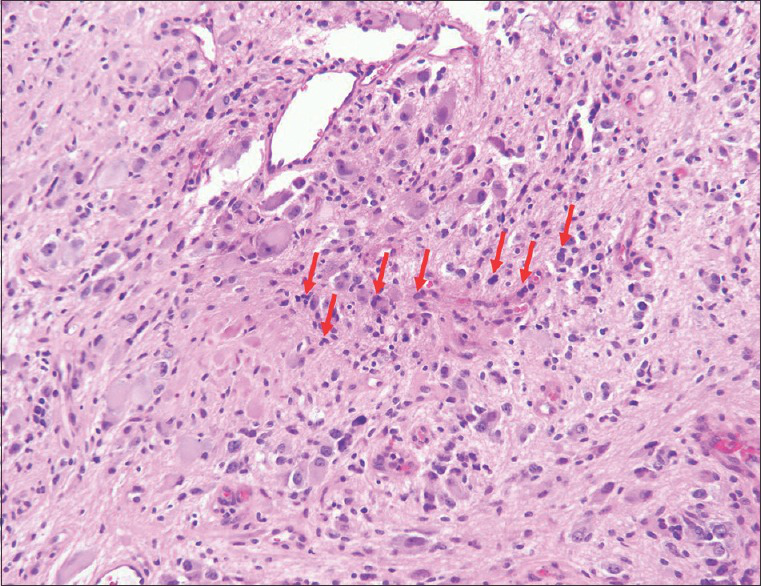
图3:4个月大的女童,患有脊髓神经节细胞胶质瘤。苏木精和曙红染色(×200)。手术切除标本显示肿瘤由许多成簇的发育异常的双核神经节细胞组成(箭头)。
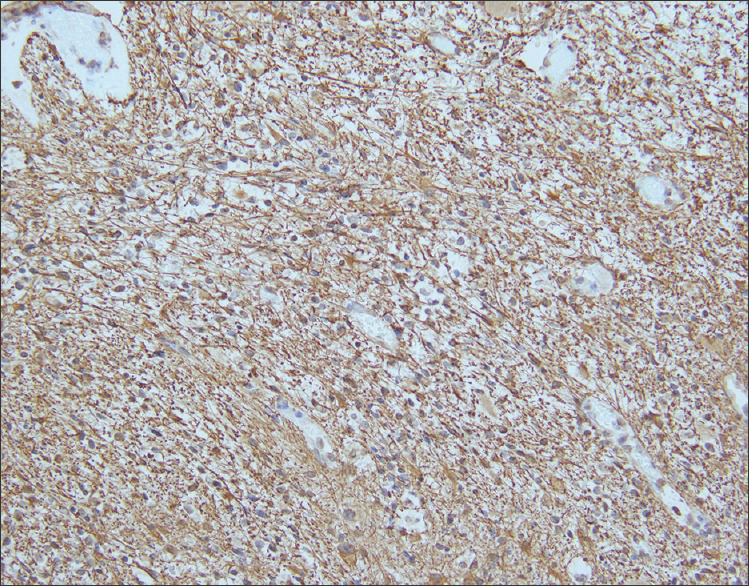
图4:14个月大的女童,患有脊髓神经节细胞胶质瘤。对手术切除标本进行的免疫组织化学显示原纤维GFAP免疫反应性星形胶质细胞(棕色)横跨肿瘤基质(用二氨基联苯胺色素和苏木精复染进行抗GFAP免疫组织化学染色;原始放大倍数×200)。
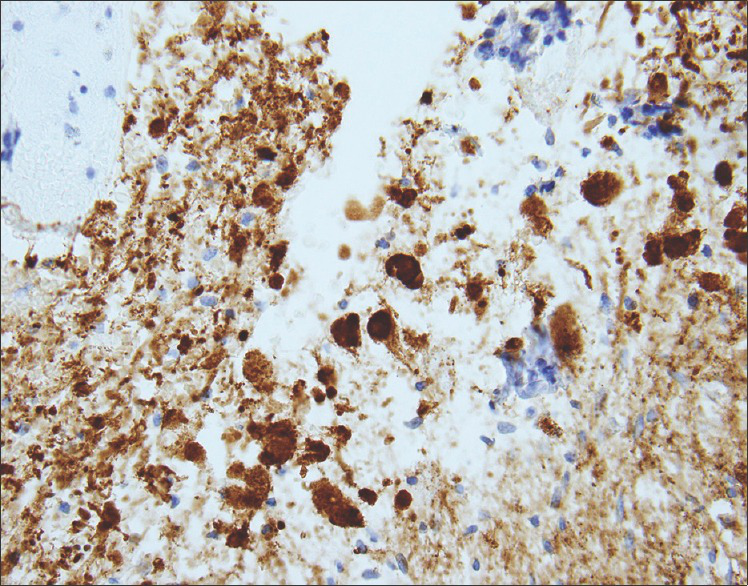
图5:14个月大的女童,患有脊髓神经节细胞胶质瘤。在手术切除标本上进行的免疫组织化学显示发育异常的双核神经节细胞,在胞体和突起中具有广泛的突触素免疫反应性(棕色)(用二氨基联苯胺色素和苏木精复染进行抗突触素免疫组织化学染色;原始放大倍数×200)。
案例总结
该例14个月大的女孩广泛的胸椎神经节细胞胶质瘤,尽管有肿瘤生长的影像学证据,其临床症状在次全切除术和辅助化疗后仍保持稳定。虽然神经节细胞胶质瘤是一种少见的脊髓肿瘤,但在髓内肿瘤的鉴别诊断中需考虑到它。早期识别、诊断和治疗对于好转预后和尽量减少神经后遗症至关重要。不幸的是,在临床实践中,由于这种肿瘤的缓慢生长和惰性,诊断经常被延迟。根治性切除术的益处需与神经功能进一步恶化的风险进行权衡,特别是在预期寿命较长的年轻患者中。有必要进一步研究部分切除和复发的脊髓神经节细胞胶质瘤的最佳治疗方法。
