



促纤维增生性婴儿节细胞胶质瘤和促纤维增生性婴儿星形细胞瘤初期被认为只发生于2岁以内,但后续有研究报道,也有25岁的患者患上该疾病。婴儿通常表现为巨头畸形以及囟门紧绷隆起。肿瘤往往大,位于大脑半球,通常兼有实性和囊性成分,在T2加权MRI上呈明显的低信号。今天就跟随脑博士一起来了解一下什么是促纤维增生性婴儿节细胞胶质瘤/促纤维增生性婴儿星形细胞瘤吧!
1、什么是促纤维增生性婴儿节细胞胶质瘤/促纤维增生性婴儿星形细胞瘤?
促纤维增生性婴儿节细胞胶质瘤(DIG)和促纤维增生性婴儿星形细胞瘤(DIA)是良性胶质神经元或胶质肿瘤,主要发生在婴儿大脑半球,由MAPK通路激活驱动,由星形细胞和神经元混合成分(DIG)或仅由星形细胞成分(DIA)嵌入广泛的促纤维增生性基质组成,通常含有未分化胚胎样肿瘤细胞病灶(CNS WHO 1级)。
2、好发部位是哪里?
SEGAs通常起源于孟氏孔区域的室管膜下结节,可以是单侧或双侧的。SEGAs是生长缓慢的肿瘤,通常在梗阻性脑积水发生前没有症状。通过系列神经影像学上增大的大小或通过梗阻性脑积水的体征和症状与室管膜下结节相区别。在没有干预的情况下,SEGAs通常会在几周到几个月内继续缓慢增长,只有很少的证据表明衰退或增长稳定。少见地,SEGAs可表现出更具攻击性的行为,与实质侵犯或广泛的周围水肿有关,或者它们可发生在非典型部位,如松果体区或下丘脑区。它们通常伸入脑室,可引起急性或慢性脑积水。
3、这种疾病有什么样的临床特征?
较常见的临床体征是头围增加、囟门隆起、嗜睡和落日征。
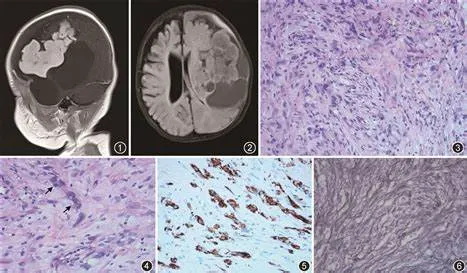
4、在影像上面有什么样的展现形式?
多见于结节性硬化症的儿童及年轻患者(平均年龄11岁)
主要症状
(1)梗阻性脑积水
(2)由于急性颅内压增高引起的头痛、恶心、呕吐等症状
(3)由于脑室扩张引起的自发性瘤内出血少见
(4)可见逐渐加重的癫痫发作
肿瘤全切后预后良好
治疗方法:雷帕霉素和/或手术切除
5、这种疾病有什么样的流行病学呢?
DIG/DIA是少见的原发性脑肿瘤,但其真实发生率尚不明确,因为它们通常包括在胶质神经元肿瘤大组中。DIG/DIA占脑肿瘤的0.4%,M:F比为1.8:1。它们占儿童颅内肿瘤的1.25%,占婴儿脑肿瘤的1.3-15.8%。多数病例发生在24月龄之前。已经报道了少见的非婴儿病例,但这些病例发生在前遗传学时代,可能代表错误分类的瘤种。
6、什么样的发病机制会造成这种疾病的产生呢?
尽管确切的细胞来源仍不确定,但推测DIG/DIA来自发育大脑中的不同软脑膜下星形胶质细胞群。然而,在大多数病例中,未分化的胚胎样肿瘤细胞病灶的存在增加了早期祖细胞产生具有不同程度进行性成熟的肿瘤的可能性。DIG/DIA在遗传学上由MAPK信号转导通路的激活驱动。
7、肉眼外观是什么样的?
DIG/DIA通常含有大的单房或多房囊肿,囊内充满透明无色或黄色液体。实性浅表部分主要在脑外,累及软脑膜和浅表皮质。肿瘤通常附着在硬脑膜上,质硬或呈橡胶状,颜色为灰色或白色。通常没有出血或坏死的大体证。
8、在组织病理学上有什么样的特征?
DIG/DIAs是由明显的促纤维增生性软脑膜间质和不同比例的神经上皮成分组成的双相性肿瘤。促纤维增生性软脑膜成分由成纤维细胞样梭形细胞与胶原基质混合组成。富含网织蛋白的基底膜通常包围几乎每个细胞。在DIA中,神经上皮成分仅包括星形胶质细胞群;在DIG中,还观察到具有神经节细胞分化的肿瘤性神经元成分。肿瘤性星形胶质细胞呈束状或席纹状或漩涡状排列。此外,DIG/DIA通常含有原始胚胎样肿瘤细胞病灶。这种不成熟的成分,缺乏结缔组织增生,可能在某些区域占优势。
皮质表面和促纤维增生性肿瘤之间有一个明显的分界,尽管Virchow–Robin间隙经常充满肿瘤细胞。钙化常见,但通常不存在血管周围单核炎性细胞浸润和黄色瘤细胞。坏死不常见,通常仅限于原始胚胎样细胞病灶。通常不存在肾小球样微血管增生。在大多数病例中,有丝分裂象仅限于胚胎样肿瘤细胞病灶,促纤维增生成分中有丝分裂象不超过0.8个核分裂/mm^2【相当于的<2个核分裂/10HPF(直径0.55 mm、面积0.24 mm^2)】。
9、其免疫组织化学有什么表现?
胶质成分强烈表达GFAP,而在肿瘤性神经节细胞以及缺乏明显神经元分化的细胞中观察到神经元标志物(突触素syn、神经丝蛋白NF、NeuN)。促纤维增生成分内的Ki-67增殖指数范围为<0.5%至5%,大多数报告值<2%。然而,Ki-67指数在胚胎样肿瘤细胞病灶中可升高(高达20%)。使用VE1克隆的免疫组织化学可用于检测携带BRAF p.V600E突变的DIG/DIA,但该突变特异性抗体不能识别p.V600D突变或在不携带p.V600E突变的DIG/DIA中发现的任何其他BRAF突变或重排。
10、如何进行鉴别诊断?
DIG/DIA的主要鉴别诊断是节细胞胶质瘤GG、多形性黄色星形细胞瘤PXA和新定义的瘤种婴儿型半球胶质瘤。与DIG/DIA一样,节细胞胶质瘤和多形性黄色星形细胞瘤通常是具有频繁BRAF突变的囊实性肿瘤,但节细胞胶质瘤和多形性黄色星形细胞瘤通常要小得多,并且发生在年长儿童中,相比之下,DIG/DIA的体积大且在婴儿中发病。婴儿型半球胶质瘤与DIG/DIA具有婴儿期发病和大脑半球位置的特征,但婴儿型半球胶质瘤通常缺乏促纤维组织增生并表现出浸润性生长模式(与DIG/DIA不同),它们的遗传学特征为涉及受体酪氨酸激酶基因(ALK、ROS1、MET、NTRK1、NTRK2和/或NTRK3)的融合。
11、有什么诊断分子病理学?
DIG/DIA是IDH和组蛋白H3野生型肿瘤,其特征是基因改变引起MAPK信号通路激活,较常见的是通过涉及BRAF或RAF1的突变或融合。BRAF突变可包括p.V600E和相同密码子的其他变异,如p.V600D,此外还包括其他位置的变异或涉及KIAA1549以外伴侣的融合。这些BRAF或RAF1突变或融合通常是确定的致病性改变,而DIG/DIA缺乏多形性黄色星形细胞瘤中通常伴随BRAF改变的CDKN2A和/或CDKN2B纯合性缺失。虽然已经报道了携带ALK或NTRK融合的组织学定义的DIG/DIA的个体病例,但在携带ALK、ROS1、MET或NTRK融合的婴儿中发生的大多数大脑半球胶质瘤在表观遗传学上与婴儿型半球胶质瘤聚集。是否存在也携带受体酪氨酸激酶基因(ALK、ROS1、MET、NTRK1、NTRK2、NTRK3或FGFR1)融合的DIG/DIA仍有待确定。DNA甲基化分析显示,DIG/DIA的表观遗传特征不同于迄今描述的全部其他原发性CNS肿瘤实体,包括多形性黄色星形细胞瘤和上述婴儿型半球胶质瘤。
12、都有什么治疗方式呢?
治疗方法为手术,在不完全切除的情况下应联合化疗。大多数研究表明,肉眼下全切可带来长期存活。通常只有在已用尽其他方法后才考虑放疗。
13、有怎么样的预后效果?
当完全手术切除时,DIG/DIA的预后较好,大多数病例随访5-15年没有复发。半球位置的长期结局优于鞍上位置,而软脑膜播散和/或多灶性疾病虽然少见,但较常与鞍上位置相关。在仅进行次全切除或活检的情况下,需进行仔细随访以监测残留肿瘤的再生长,通常情况下,残留肿瘤会在数年内保持稳定或缓慢生长,但也有记录显示肿瘤消退。在次全切除病例中,辅助化疗和/或放疗是一种治疗性考虑,是对于残留肿瘤进展和/或软脑膜播散的患者。靶向MAPK信号转导通路的小分子酪氨酸激酶控制剂也是BRAF突变病例的治疗选择。尽管一些DIG/DIA显示了明显间变的病灶(例如,高有丝分裂计数、栅栏状坏死),但间变与临床结局之间没有明确的关系。一次切除后8~10年复发并伴有DIG/DIA恶变的少见病例已有报道,个别病例还伴有获得性TP53、ATRX或BCORL1突变。
14、长期管理是怎么样的情况呢?
尽管在组织学上观察到恶变,但一些患者在二次手术后仍存活≥3年。
