



视觉系统肿瘤发生在许多遗传性疾病中,包括视网膜母细胞瘤RB1基因(沃格尔1979年),结节性硬化症中的视网膜星形细胞错构瘤(罗利等人,2001年)、冯-希佩尔-林道病视网膜血管母细胞瘤(Lonser等人,2003年)、神经纤维瘤病2型中的视神经鞘脑膜瘤(Bosch等人,2006年),以及1型神经纤维瘤病(NF1)(李斯特尼克等人,2007年).在这些疾病中,NF1是较常见的,在范围内影响1/3000的个体(克劳1956;埃文斯等人,2010年;弗里德曼1999年;Huson等人,1989年).
NF1是一种常染色体显性综合征,由染色体中的功能缺失(LOF)突变引起NF1肿瘤控制基因(Cawthon等人,1990年;Viskochil等人,1990年;华莱士等人,1990年),它编码神经纤维蛋白。神经纤维蛋白由2800多个氨基酸(~220 kDa)组成,包含一个小结构域(280-300个氨基酸),在结构和功能上类似于作为负性RAS调节因子的蛋白质家族(Basu等人,1992年;Bollag和McCormick 1991年;徐等1990a;徐等1990b).与RAS激活增加与多种人类癌症相关联相一致(帝国等2017;西曼舒等人,2017年),NF1患者易患一系列影响中枢和外周神经系统的肿瘤,包括OPGs,这是该人群中发病率的来源(李斯特尼克等人,2007年).
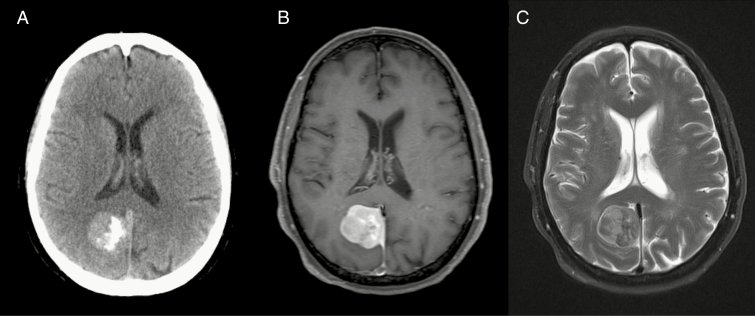
几乎全部的NF1相关OPGs(NF1 OPGs)都是良性毛细胞星形细胞瘤(PAs国际卫生组织I级星形细胞瘤),其可以出现在沿视神经通路的任何地方,包括视神经、视交叉、视束和视放射(Guillamo等人,2003年;李斯特尼克等人,2007年;刘等2004)(图1A–C).在患有NF1的个体中,大多数(75-85%)OPGs位于视神经和视交叉内(视交叉前或前视通路),较小比例的肿瘤位于视束和放射区(视交叉后或后视通路)。NF1-OPGs较常发生在幼儿(诊断时的中位年龄=4.5岁)(李斯特尼克等人,1994年;李斯特尼克等人,1989年;普拉达等人,2015年),在年龄较大的青少年中描述了少见的病例(Chong等人,2013年;李斯特尼克等人,2004年).因此,OPG是NF1的一种表现,其在幼儿中占主导地位,这些幼儿通常患有共病注意力缺陷,这使得在高危人群中的诊断和准确视觉评估更加复杂(李斯特尼克等人,2007年).
NF1-OPG在它们的位置、初次检测的年龄和视力丧失方面表现出的临床异质性(李斯特尼克等人,1994年).NF1-OPG病导致视力丧失的危险因素是年龄小于2岁(费希尔等人,2012年),女性性别(Diggs-Andrews等人,2014年b),以及视交叉后视神经通路的肿瘤累及(Balcer等人,2001年;费希尔等人,2012年).治疗通常保留给具有进行性症状的患者(例如。视力丧失)并经常涉及化疗。虽然大多数有症状的儿童用卡铂/长春新碱治疗(Mahoney等人,2000年;帕克等人,1997年;Packer等人1993年),分子靶向治疗近期出现了。化疗通常能成功控制肿瘤生长(60-70%的反应率),但是很少有患者在治疗后视力得到好转(Dalla Via等人,2007年;道奇顺等人,2015年;费希尔等人,2012年;李斯特尼克等人,2007年;Shofty等人,2011年).
大多数接受化疗的患者没有先前的组织诊断(即。肿瘤活组织检查),并且很少有患者接受其肿瘤的手术切除。缺乏用于研究的人类肿瘤标本和患者来源的异种移植物模型阻碍了理解人类NF1-OPG的分子和细胞决定因素以及发现新的治疗方法的努力。为此,我们对NF1-OPG病的了解大多来自于对小鼠的研究Nf1基因(Gutmann等人,2012年).在这篇综述中,我们总结了我们目前对NF1-OPG病理生物学的理解,包括改造成港湾型的小鼠视力丧失的机制Nf1视神经胶质瘤。我们进一步讨论了与预防或限制进行性视力丧失相关的分子靶向和神经保护疗法的发展前景。
参考资料: J Neurosci Res.2019 Jan;97(1):45–56.
Published online 2018 Apr 28.doi:10.1002/jnr.24250
